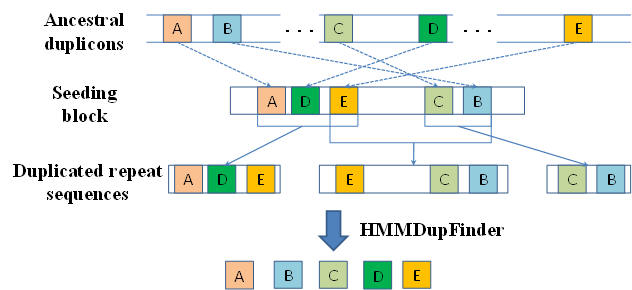
HMMDupFinder: A Framework for Finding Duplicons
Given a set of large repeat sequences (e.g., segmental duplications and copy number variations), the HMMDupFinder aims to identity the small subrepeat unis (termed duplicons) within the large repeats. The developed programs within HMMDupFinder are implemented in C++ and encapsulated via shell script on Linux, which are freely available for academic use.
-
Installation
-
Download HMMDupFinder
- HMMDupFinder.tar.gz (binary version on 32-bit Linux, which can be directly run without compilation)
- The Source code can be
downloaded and compiled for other platforms (with g++ 4.3.5 or higher).
-
Unpack Distribution
- Unpack the distribution on your Linux server (e.g., /home/myself/) via
- tar -zxvf HMMDupFinder.tar.gz
- cd HMMDupFinder
- The HMMDupFinder folder should contain two main scripts:
HMMDupFinder.sh and PermutationTest.sh, which
encapsulate 11 programs of the entire framework, and two
test files (TestRepeatFile and TestBLASTFile).
-
Run the HMMDupFinder
- The HMMDupFinder.sh script (bash) aims to identify all potential duplicons using the proposed HMM framework. To run HMMDupFinder, simply type
- ./HMMDupFinder.sh
Parameter1 Parameter2
- Running this script requires two input parameters.
- Parameter 1: Coordinates of the repeat sequences (see an input example TestRepeatFile).
- Parameter 2: Processed BLAST alignment result (see an input example TestBLASTFile and the preprocess section)
- e.g., using the provided input repeat
file (TestRepeatFile) and alignment file (TestBLASTFile),
simply type:
sh HMMDupFinder.sh TestRepeatFile TestBLASTFile
- Running this script requires two input parameters.
- This script will create a folder 'Duplicon'
which stores the coordinates (chr, start, end) of potential duplicons
within each repeat sequence. e.g., there will be
two duplicons found in the 3_100_1000 repeat.
3 249 349
3 499 649
- The PermutationTest.sh script will perform permutation test to assess the significance of identified duplicons. Only duplicons with statistical significance (P<0.01) will be outputted. Note that the permutation may take longer time as the repeat sequences are very large.
- ./PermutationTest.sh Parameter1 (Parameter1 refers to the repeat sequence file same as the above)
- The coordinates of duplicons (chr, start, end) passing the permutation
test will be stored in the 'Duplicon_After_Test/' directory.
-
-
Preprocess of BLAST Alignments
- The HMMDupFinder requires input of BLAST tabular alignments (i.e., -m8 tabular format) among the duplicated sequences (http://www.ncbi.nlm.nih.gov/blast/download.shtml).
- Please choose one set of repeat sequences (e.g., CNVs) as
the subject database and the remaining duplication sequences
(e.g., segmental duplications) as queries during BLAST
alignment.
- e.g., megablast -d CNV.fa -i chr1_SD.fa -m8 -e 0.00001 -W 36 -o BLASTResult_chr1 -F F
- Convert the BLAST tabular format into the the input format
required by HMMDupFinder (see an input
example).
- Format: the first six fields stand for the
coordinates of two repeat sequences, and the remaining four
fields stand for the BLAST results of pairwise local alignments,
separated by white space.
<Repeat1_Chr> <Repeat1_Start> <Repeat1_End> <Repeat2_Chr> <Repeat2_Start> <Repeat2_End> <Alignment Identify> <Length> <Repeat1_Alignment_Start> <Repeat1_Alignment_End> <Repeat2_Alignment_Start> <Repeat2_Alignment_End> - If the coordinates of your sequences are put on the fasta header as sequence IDs (e.g., >3 100 1000), you can simply convert the blast output into the required format by typing "cut BLASTResult_chr1 -f 1-4,7-10 | tr '\t' ' ' > TestBLASTFile"
- Format: the first six fields stand for the
coordinates of two repeat sequences, and the remaining four
fields stand for the BLAST results of pairwise local alignments,
separated by white space.
Genomics Research Center, Academia Sinica, Taiwan.
Department of Computer Science and Information Engineering, National Chung
Cheng University, Taiwan.
For any question, please contact
Trees-Juen Chuang: trees@gate.sinica.edu.tw, or
Yao-Ting Huang: ythuang@cs.ccu.edu.tw